Deucravacitinibと2つのメチル基
久々に書く気になったのでこちらの続きです。
APIで取得したChEMBLのデータを整える - おじさんちのクソゲ
Deucravacitinibは2022.9に承認されたTYK2に対するアロステリック阻害剤で、成人の中等度から重症の尋常性乾癬を適応としているそうです。
なんでも、JH2特異的に結合することでkinase活性をmodulateするんだとか。
JH2ってなに?
the JAKs possess two near-identical phosphate-transferring domains. One domain exhibits the kinase activity, while the other negatively regulates the kinase activity of the first. Janus kinase - Wikipedia
とのことで、リン酸転移ドメインは二つあるけど使ってるのは一個で、2個目のJH2はフェイクってことみたいです。 DeucravacitinibはフェイクのJH2に結合するにも関わらず阻害活性を示すことから、アロステリック阻害剤ということみたいです。
先日の検索結果から新しいものを少し読んでみると、面白い内容でした。
https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.9b00444
残念ながらフリーでは読めませんでしたが、孫引きまで含めて概要は下記の通りでした。
1. BMSの社内ライブラリ由来
2. cell baseのSPAで周辺化合物を評価してユニークな阻害活性を見出す
3. 活性がないはずのJH2に対して強くbindingしていた
4. メチル基1個でJAK1~3, TYK2のJH1阻害活性が完全に切れた(!) 一つ目のメチル
5. 正確なMoAは不明だが、実験的な事実としてDeucravaciinibはTYK2の不活性化状態を安定化してるっぽい (間違ってたらごめんなさい)
6. catalityc pocketの向きに必要な置換基をメチルスルホン > メトキシ 二つ目のメチル
7. 代謝された脱メチル体は選択性が低いことから毒性懸念浮上 (それでもぱっと見は他よりよい)
8. D化メチル体によって代謝物抑制
9. 母核変換でPK改善
というわけで候補品に至ったようです。ちなみに1で得た他の骨格を進めたNimbus子会社を武田が買ったとかなんとか。
自己免疫疾患に新規作用機序の「TYK2阻害薬」―ブリストルが乾癬で承認取得、武田は買収で参入 | AnswersNews
ちなみにDeucravacitinibは重水素が3つも入ってるんですが、ケミストなら代謝安定性改善したかったのかな?とか考えちゃいます。
私自身トライしてみて全く改善したことがなかったのですが、特定の代謝物を抑制する効果はみられるんだとか。
親化合物のPKは改善しませんでしたが、代謝物を抑制した経験はあります。代謝パス変わるんで、毒性抑えるという使い方はありかもですね!
— へい🍅 (@HiGoing) December 21, 2022
確かに普段の評価では化合物の残存を見ているわけですが、望まない副生物をきちんと抑えることができれば目的を達成できることもあるわけです。
BMSの報告には正にその記述がありました。Deuterium kinetic isotope effectというらしいです。
https://pubs.acs.org/doi/10.1021/bi972266x
上記以外にもアミドのHBDを減らすためのヘテロ環への変換や、置換基探索で芳香環をc-Pr基に変換できたことへの考察など、探索から最適化までのエッセンスが詰まった全体の仕事となっています。久しぶりにメドケムが仕事してんな!っていう論文を読んだ気がしました。
やる気が出たら、次回データ解析後編に続きます。。
Dockerfile@M1 Mac突貫工事
最初のFROM --platform=linux/amd64 ubuntu:latestに集約されますが、M1 MacでDockerfileを単にbuildしようとすると、minicondaのインストールでエラーが出ました。
というわけでとりま突貫のdockerfileです。
拡張子なしでcurrent directoryに保存して、下記コマンドでdocker imageにbuildできます。
docker build -t `image_name` .
こちらがdockerfileです。
# M1 MacでUbuntuコンテナを作る時の注意 # https://qiita.com/silloi/items/739699337b9bf4883b3e FROM --platform=linux/amd64 ubuntu:latest # update RUN apt-get -y update && apt-get install -y \ libsm6 \ libxext6 \ libxrender-dev \ libglib2.0-0 \ sudo \ wget \ unzip \ vim #install miniconda3 WORKDIR /opt # download miniconda package and install miniconda # archive -> https://docs.conda.io/en/latest/miniconda.html RUN wget https://repo.anaconda.com/miniconda/Miniconda3-latest-Linux-x86_64.sh RUN bash /opt/Miniconda3-latest-Linux-x86_64.sh -b -p /opt/miniconda3 RUN rm -f Miniconda3-latest-Linux-x86_64.sh # set path ENV PATH /opt/miniconda3/bin:$PATH # conda create RUN conda create -n pymol python=3.7 # install conda package SHELL ["conda", "run", "-n", "pymol", "/bin/bash", "-c"] # https://qiita.com/kimisyo/items/66db9c9db94751b8572b RUN command conda config --append channels conda-forge RUN conda install -c conda-forge rdkit -y RUN conda install -c conda-forge pymol-open-source -y RUN conda install plotly RUN conda install -c conda-forge nodejs jupyterlab RUN conda install -c conda-forge py3dmol WORKDIR / RUN mkdir /work WORKDIR / RUN conda init # かめさんのdocker image_name # datascientistus/ds-python-env # ubuntuというdocker imageをbuild # docker build -t ubuntu . # 最初からbuildするなら--no-cache # docker build -t ubuntu . --no-cache # sbddというcontainerをubuntuというimageからcreateしてbashでrun # docker run -it -v ~/Documents/Linux:/work --name sbdd ubuntu bash
私の環境では最後のRUN conda initがないと、コンテナでcondaコマンドを使う時に下記のエラーが出ました。
CommandNotFoundError: Your shell has not been properly configured to use 'conda activate'.
To initialize your shell, run
$ conda init <SHELL_NAME>
Currently supported shells are:
- bash
- fish
- tcsh
- xonsh
- zsh
- powershell
See 'conda init --help' for more information and options.
IMPORTANT: You may need to close and restart your shell after running 'conda init'.
かめさんのUdemy講座では下記のscriptを追加して、bashを起動せずにhost側のlocalhost:8888からコンテナにアクセスしてました。
# execute jupyterlab as a default command CMD ["jupyter", "lab", "--ip=0.0.0.0", "--allow-root", "--LabApp.token=''"] # ubuntuというimageからsbddというコンテナをポート8888に繋いでrun # docker run -v ~/Documents/Linux:/work -p 8888:8888 -it --name sbdd ubuntu
この方法だとPCの再起動なんかでやむを得ずコンテナを止めてしまった時に、restartしてもlocalhost:8888からjupyterに入れませんでした。
気になるので問い合わせ中です。
Dockerを猛復習
Dockerの全体像
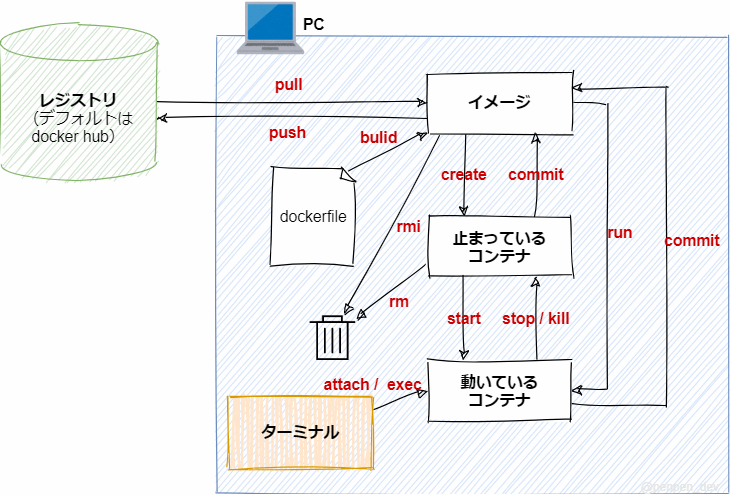
https://penpen-dev.com/blog/docker-command/
出力される結果は各自の環境で異なるので参考程度にしてください。
Contents
新しい環境を構築する
docker hubからimageをpull
docker pull `image:ver` docker pull ubuntu # ver.を指定しないとlatestがpullされる
pullしたimageを参照
docker images # REPOSITORY TAG IMAGE ID CREATED SIZE # ubuntu latest 4c2c87c6c36e 7 weeks ago 69.2MB # python latest 9dda5bedc1c7 3 months ago 868MB
imageからコンテナを作ってrun
enterはしない
imageを持ってない場合はhubからpullしてくる
docker run `image_name` docker run --name `container_name` `image_name` # --name ``でcontainer nameを指定できる docker run --name test hello-world # Hello from Docker! # This message shows that your installation appears to be working correctly. # ...
imageからcontainerを作ってbashで作業する
docker run --name `container_name` -it `image_name` bash # interact terminal # root@container_id:/# # containerのbash promptが返ってくる root@container_id:/# exit # containerを停止してexitする # directory$ # hostのterminal promptに戻る
imageからcontainerにlocal volumeをマウントしてbashで作業する
docker run --name `container_name` -v `host abs. path`:`container abs. path` `image_name` `command` docker run --name `container_name` -v ~/Documents/Linux:/work -it ubuntu bash
imageからcontainerにlocal volumeをマウントしてjupyterのポートを割り当ててbashで作業する
docker run -v ~/Documents/Linux:/work -p 8888:8888 --name test ubuntu # container側にJupyterなどが入ってないとhostのブラウザで`localhost:8888` を入力してもjupyterは起動しない
Containerで作業する
containerのリストを表示
docker ps # upのものだけ docker ps -a # 全て表示 # CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES # 498900d17e03 hello-world "/hello" 2 minutes ago Exited (0) 2 minutes ago test # cdf417f58458 continuumio/anaconda3:latest "/bin/bash" 3 months ago Exited (137) 20 hours ago anaconda3 # runしただけではcontainer statusはexited
containerを起動する
docker restart `container_name` docker restart test # test docker ps -a CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES 498900d17e03 hello-world "/hello" 7 minutes ago Exited (0) 48 seconds ago test cdf417f58458 continuumio/anaconda3:latest "/bin/bash" 3 months ago Exited (137) 20 hours ago anaconda3 # hello-worldにはOSとbashが入ってないので起動(=up)にならない
docker restart anaconda3 # anaconda3 docker ps # status == up のcontainerを表示 # CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES # cdf417f58458 continuumio/anaconda3:latest "/bin/bash" 3 months ago Up 5 seconds anaconda3
起動中のcontainerに入って新しいbashで作業する (execute)
docker exec -it `container_name` bash # root@container_id:/# # containerのbash promptが返ってくる
docker attachとの違いについて
https://qiita.com/RyoMa_0923/items/9b5d2c4a97205692a560
attach : container内の既に起動しているbashを使う
exec -it : 新たにbashを立ち上げる
起動したままcontainerから抜ける (detach)
ctrl+p ctrl+q
VS codeではVS codeのショートカットが起動してしまうので変更する必要がある
https://qiita.com/Statham/items/c204e85067ea4dca2724
docker ps # CONTAINER ID IMAGE COMMAND CREATED STATUS PORTS NAMES # cdf417f58458 continuumio/anaconda3:latest "/bin/bash" 3 months ago Up 10 minutes anaconda3
起動中のcontainerのbashに入って作業する
docker attach `container_name` # root@container_id:/# # container/bashのpromptが返ってくる
containerを停止して脱出する
root@container_id:/# exit docker ps -a STATUS PORTS NAMES # cdf417f58458 continuumio/anaconda3:latest "/bin/bash" 3 months ago Exited 10 seconds anaconda3
環境を破棄する
Containerはimageを基に動いているので、containerが存在するうちはimageを削除できない。
手順としては
1. containerをimageにcommit
2. imageをdockerhubにpush
3. containerを削除
4. imageを削除
で環境を保存しながら適宜破棄する。
もしくはDockerfileとして残す。
コンテナをimageにcommit
docker commit `container_name` `image_name:ver` docker images # REPOSITORY TAG IMAGE ID CREATED SIZE # ubuntu latest 4c2c87c6c36e 7 weeks ago 69.2MB # python latest 9dda5bedc1c7 3 months ago 868MB # continuumio/anaconda3 latest 40d5cbe3a8cd 7 months ago 2.69GB docker commit anaconda3 continuumio/anaconda3 # sha256:1b5cfa5999b6dceb07ddd5ebea6c7c24ec98d61a783fa719443a5adb2a4016b9 docker images # REPOSITORY TAG IMAGE ID CREATED SIZE # continuumio/anaconda3 latest 1b5cfa5999b6 4 seconds ago 3.26GB # ubuntu latest 4c2c87c6c36e 7 weeks ago 69.2MB # python latest 9dda5bedc1c7 3 months ago 868MB # continuumio/anaconda3 <none> 40d5cbe3a8cd 7 months ago 2.69GB
docker_hubにログイン
docker login # idとpwを入力
imageをdocker hubのrepoにpush
docker push `user_id/image_name:ver`
containerを削除
docker rm `container_name` # or `container_id`
imageを削除
docker rmi `image_name` docker rmi continuumio/anaconda3 docker images # REPOSITORY TAG IMAGE ID CREATED SIZE # ubuntu latest 4c2c87c6c36e 7 weeks ago 69.2MB # python latest 9dda5bedc1c7 3 months ago 868MB # continuumio/anaconda3 <none> 40d5cbe3a8cd 7 months ago 2.69GB # 指定しないとlatestが消えるっぽい # latestがないimageは`:ver`でtagを指定しないと消えない # containerで使用中だとreferenceになっているため削除できない
dockerfileからimageをbuild
cd `working directory` docker build -t `image_name` . docker build -t `image_name` . --no-cache # オプションないと前回の続きをbuildする
Reference
Docker超入門
ぷんたむの悟りの書
https://punhundon-lifeshift.com/dockerfile
miniconda + conda-forgeで開発環境を揃える
ChEMBLのデータを可視化してみた
前回までのこちらの記事でChEMBLからダウンロードしたデータを可視化できるように加工しました。
既に構造や骨格の情報となるMol fileをSMILES stringsから生成してるので、今回は続きとして同じDataFrameを使っていきたいと思います。
可視化によく使われるのはmatplotlibとかseabornですが、今回はplotlyを使ってます。
簡単にintractiveなグラフを書いたり、web pageに組み込んだりできる(らしい)ので試してみたかっただけです。
- 統計値の作成
- Count of Documents and Scaffold by Year
- pChEMBL Value box by Year
- Filtering by Molecular Name
- Molecule Max Phase scatter by Year
- Summary Table of Deucravacitinib
統計値の作成
Spotfireとかだと文献や骨格情報のUnique countとかを簡単に表示できるやつです。
他にいい方法があったら教えてください。
dfd = df.groupby(['Document Year'])['Document ChEMBL ID'].describe() dfm = df.groupby(['Document Year'])['Murcko_generic_SMILES'].describe() dfd

Document Yearをgroupbyでまとめた後、Document ChEMBL IDとMurcko_generic_SMILESの統計値をとってあげたっていう感じです。
注意が必要なのは、前回の処理でDocument Yearが不明なものを2025として処理していることです。
0とかでもいいんですが、可視化した時の見やすやを重視しています。
Count of Documents and Scaffold by Year
plotlyを使うとこんな感じです。
import plotly.graph_objects as go fig = go.Figure() fig.add_trace(go.Bar(x=dfd.index, y=dfd['unique'], name='Document count')) fig.add_trace(go.Bar(x=dfg.index, y=dfg['unique'], name='Scaffold count')) fig.add_trace(go.Bar(x=dfm.index, y=dfm['unique'], name='Scaffold count')) fig.update_layout( title='Documents and Murcko scaffold v.s. Year', # showlegend=False, # Legendのon/off width=1000, # height=700 barmode='group', ) fig.show()
 Murcko Scaffoldは環構造に基づく骨格分類です。genericにすると芳香環やヘテロ環の情報もフラットにしてくれます。cyclopropylやcyclobutylが置換基ではなく骨格扱いになるところは厄介ですが、状況に応じて使い分けるかなと思います。
Murcko Scaffoldは環構造に基づく骨格分類です。genericにすると芳香環やヘテロ環の情報もフラットにしてくれます。cyclopropylやcyclobutylが置換基ではなく骨格扱いになるところは厄介ですが、状況に応じて使い分けるかなと思います。
例えばSARで置換基の一つとしてPhみたいなのが登場することを考えると、文献一報あたりの骨格数は2~5くらいじゃないかなと思います。
その観点で見ると、2011年や2015年は突出してるように見えますね。骨格変換による何かしらのブレイクスルーがあったか、FBDDやVirtual Screeningのように大規模な探索が行われたのかもしれません。
pChEMBL Value box by Year
import plotly.express as px fig = px.box( df, x='Document Year', y='pChEMBL Value', points='all', ) fig.update_layout( title='pChEMBL Value v.s. Year', showlegend=False, # Legendのon/off width=1000, # height=700 ) fig.show()

pChEMBL ValueはpIC50みたいなものと思っていいと思います。
ただし、functional assayだったりbinding affinityだったりと、各文献で評価系が異なる点に注意が必要です。
細かいところはDataFrameのassay discriptionで確認できます。
比較的早い段階からpChEMBL Value = 9が報告されていますが、TYK2はkinaseなので非選択的な既知阻害剤だったりで試験してたりするんじゃないかなと思います。
points = 'all'とすることでデータ数がわかりますが、2010年くらいから選択的阻害剤の探索が開始され、2019年くらいから選択性のいい化合物のSARが報告され出してるのかなーと妄想します。
Filtering by Molecular Name
import plotly.express as px fig = px.box( df[df['Molecule Name'].notna()], #ここを変えただけ x='Document Year', y='pChEMBL Value', points='all', ) fig.update_layout( title='pChEMBL Value v.s. Year', showlegend=False, # Legendのon/off width=1000, # height=700 ) fig.show()

治験だったり承認されたりすると薬剤名が登録されます。Molecule Nameでfilteringするとこんな感じです。
考察が正しいかはさておき、2011年くらいにやたら評価数が増えています。kinase標的の上市品が増えたとか、イベントでもあったんでしょうか?
Molecule Max Phase scatter by Year
import plotly.express as px fig = px.scatter( df[df['Molecule Name'].notna()], x='Document Year', y='pChEMBL Value', color='Molecule Max Phase', hover_name='Molecule Name', # hover_data=['Document ChEMBL ID', df[df['Molecule Name'].notna()].index], opacity=0.5 ) fig.update_layout( title='pChEMBL Value v.s. Year', # showlegend=False, # Legendのon/off width=1000, # height=700 ) fig.show()
 使われた薬剤の治験進行状況をプロットするとこんな感じでした。
使われた薬剤の治験進行状況をプロットするとこんな感じでした。
承認されたDeucravacitinibの初登場は2019年でした。いいもん出来たから文献に報告したんでしょうね。
ちなみにplotlyはlegendをダブルクリックすると簡単にfilteringできます。plotをドラッグすることで拡大したりもできます。

やはり早い段階から上市薬(Molecule Max Phase = 4)で試験されてますね。
Summary Table of Deucravacitinib
name = input('Molecule Name?') df[df['Molecule Name']==name].filter(items=[ 'Molecule Name', 'Molecule ChEMBL ID', 'Molecule_Image', 'Molecular Weight', 'pChEMBL Value', 'AlogP', 'Ligand Efficiency LLE', '#RO5 Violations', 'Assay Description', 'Document ChEMBL ID' ]).head(1)
 Deucravacitinibはこんな顔でした。(小さくてごめんなさい)
Deucravacitinibはこんな顔でした。(小さくてごめんなさい)
重水素が3つ入ってますね。
ピリダジン周りがヒンジバインダーだと思うんですが、あんまり見たことない顔かもなーって思いました。
SBDDの文献も見つかったので、そのうち紹介したいと思います。
続く。。。
APIで取得したChEMBLのデータを整える(2)
前回紹介した方法はほぼほぼコピペでした。
.only() で取得する項目を限定していましたが、全データを取得しようとするとどうなるでしょう?
activities = new_client.activity.filter(
target_chembl_id__in='CHEMBL3553',
pchembl_value__gte=5,
assay_type='B',
)
pd.DataFrame(activities).columns
Index(['activity_comment', 'activity_id', 'activity_properties',
'assay_chembl_id', 'assay_description', 'assay_type',
'assay_variant_accession', 'assay_variant_mutation', 'bao_endpoint',
'bao_format', 'bao_label', 'canonical_smiles', 'data_validity_comment',
'data_validity_description', 'document_chembl_id', 'document_journal',
'document_year', 'ligand_efficiency', 'molecule_chembl_id',
'molecule_pref_name', 'parent_molecule_chembl_id', 'pchembl_value',
'potential_duplicate', 'qudt_units', 'record_id', 'relation', 'src_id',
'standard_flag', 'standard_relation', 'standard_text_value',
'standard_type', 'standard_units', 'standard_upper_value',
'standard_value', 'target_chembl_id', 'target_organism',
'target_pref_name', 'target_tax_id', 'text_value', 'toid', 'type',
'units', 'uo_units', 'upper_value', 'value'],
dtype='object')
いっぱいカラムありますよね。残念ながらAlogPとかがないことがわかります。
ligand_efficiency はLEかな?
pd.DataFrame(activities).loc[:4, ['ligand_efficiency']]
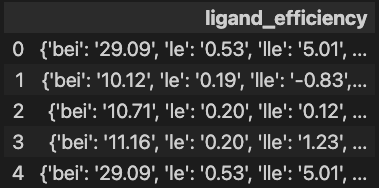
え?valueじゃなくてdictなの?
っていうことでこのままではplotや計算に使うことが出来ませんでした。What a fxxk !!
と、いう訳でpd.DataFrame()ではなくpd.json_normalize()をつかってネスト(入れ子)になったdictを各カラムに分解してやります。
df = pd.json_normalize(activities) df.iloc[:4, -4:]
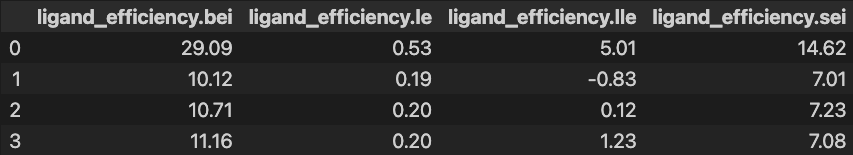
というわけで無事にAPIでDLしたデータを解析できる状態に出来ました。
扱ってみた印象だけならAPIよりcsvでDLした方が軽くて扱いやすい気がしました。
次回へ続く。
APIで取得したChEMBLのデータを整える
皆さんは標的の情報ってまず何で調べますか?
私は大体UniProtから入ることが多いです。
そして例えばUniProtのEntryをキーワードにTarget ChEMBL IDを探すこともできます。
前回みたいにChEMBLから入ることって実はあんまりありませんでした。
こちらからパクったコードにUniProt IDを入力して
# import the libraries # https://github.com/chembl/notebooks/blob/main/ChEMBL_webresource_client_examples.ipynb from chembl_webresource_client.new_client import new_client import requests import pandas as pd import platform uniprot_id = input('UniProt ID?') # P29597 target = new_client.target.get( target_components__accession=uniprot_id ).only( 'target_chembl_id', 'organism', 'pref_name', 'target_type' ) pd.DataFrame.from_records(target)

Target ChEMBL IDを指定するとあら不思議、最新版のDataをダウンロードできます。
ID = input('Target ChEMBL ID?') # CHEMBL3553 activities = new_client.activity.filter( target_chembl_id__in=[ID], pchembl_value__gte=5, assay_type='B', ).only([ 'molecule_chembl_id', 'molecule_pref_name', 'target_chembl_id', 'target_pref_name', 'parent_molecule_chembl_id', 'pchembl_value', 'canonical_smiles', 'assay_description', 'document_chembl_id', 'document_journal', 'document_year', ]) df = pd.DataFrame(activities) df[df['molecule_pref_name'].notna()].filter(items=[ 'molecule_pref_name', 'Molecule', 'pchembl_value' ] ).head(3)
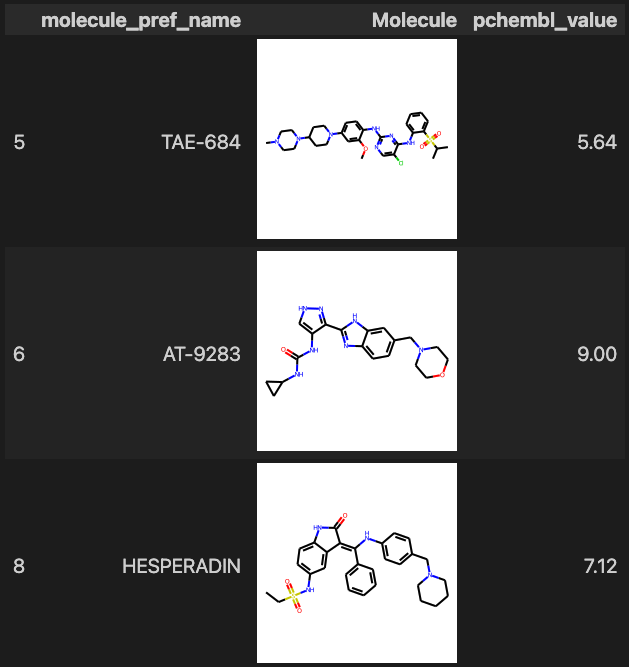
標的を複数調べる時にローカルにいちいちダウンロードするのがめんどい時はこちらの方が楽です。
KNIMEと同じルーチン化が良ければオススメです。
ただ、ダウンロード版より計算値のカラムが少ないので、同じ解析をしようと思うと自分でいくらか計算しないといけません。
あとダウンロードの時間はやっぱりかかります。一長一短ですね。
ChEMBLのデータを整える
皆さんは長期休暇の宿題は計画通り進める派でしたか?先にやる派?後でまとめて派?
私はというと、宿題とかない小学校でした笑
とは言っても子どもの学校は宿題があるので、どう接するのが正解なのかいつも悩ましいです。
今回はジジババの家に行く前に全て片付けてくれたので大満足の出だしでした。
と言うわけで?次は大人の番です(え?
先日JAKiの紹介したとこでしたが、
JAKiってキナーゼのわりには構造がシンプルでも活性と選択性が出てそうで、ケミストはとっつきやすいのかもですね!
— へい🍅 (@HiGoing) July 3, 2022
大きい買収があったりでほんと開発が活発なんだなーとか思わせるニュースを目にしました。
TYKって結構前のターゲットだった気がしてたんですが、JAKの仲間だったんですね(遅。
kinaseは各社の積み重ねが身を結んだような報告をよく目にします。
ケミストは確かにとっつきやすいのかもなーと思いますが、これからとっつく余地がどれだけあるかが創薬研究では重要かなと思います。
と言う名目で、オープンデータベースのChEMBLを使ってリガンドの外観を捉えてみたいと思います。
- Targetを検索して化合物データをダウンロード
- Import Python Libraries and Load ChEMBL Data File
- Data Cleansing
- 構造式と骨格情報の追加
- 文献数と骨格情報のメモ
- Descriptorを追加
- Summary of Data Table
Targetを検索して化合物データをダウンロード
今回はChEMBLでシンプルにTYK2を検索しました。
8 Targetsの中から一番上のCHEMBL3553を選択します。
 下の方にあるLigand Efficienciesの
下の方にあるLigand Efficienciesの
See all bioactivities for target CHEMBL3553 used in this visualization
っていう項目をクリックしてBrowse Activities を開きます。
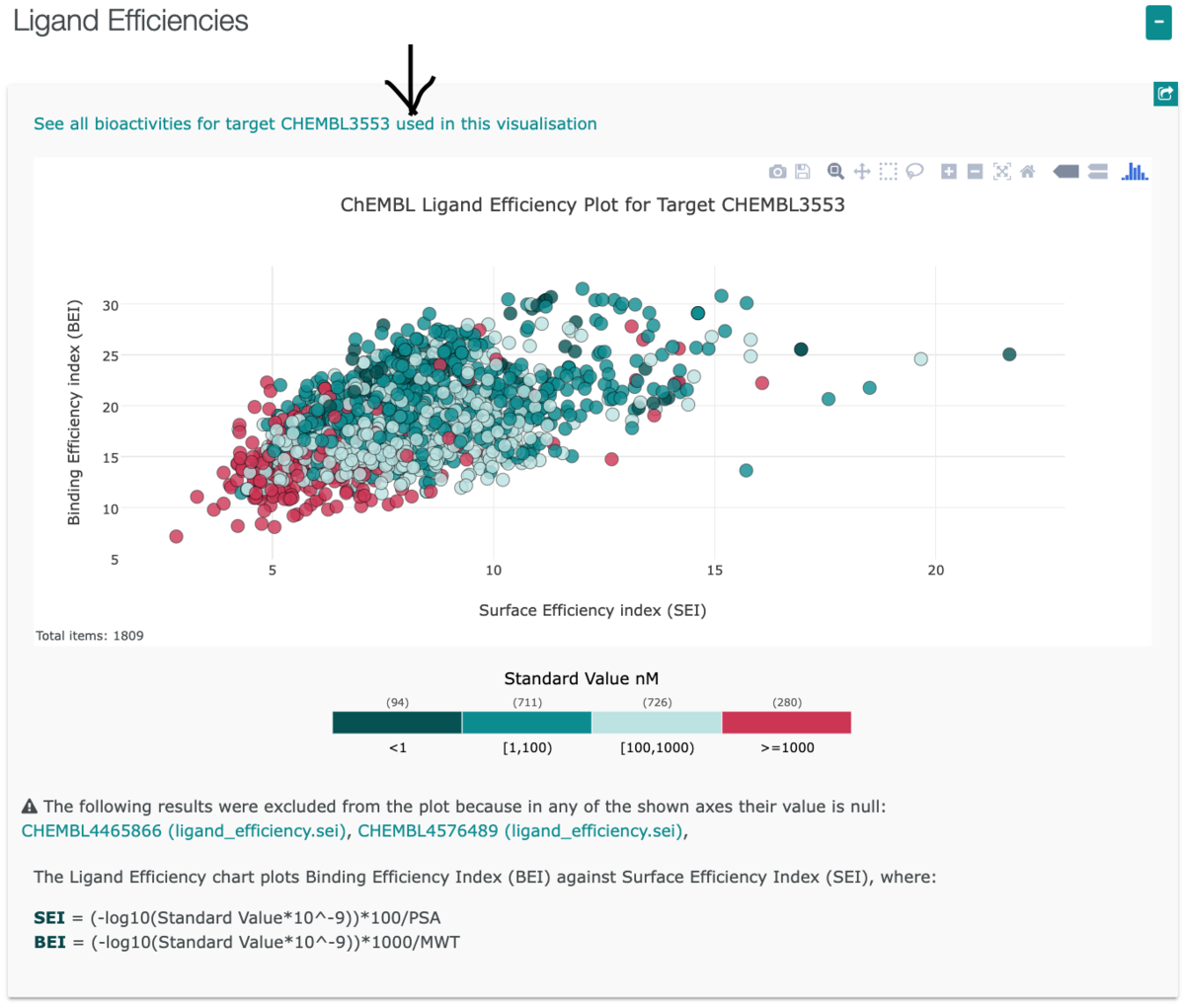 右の方にあるCSVのボタンをクリックすると化合物と活性情報のcsv fileをダウンロードできるようになります。
右の方にあるCSVのボタンをクリックすると化合物と活性情報のcsv fileをダウンロードできるようになります。
2022.12.27現在1,811 recordsでした。

Import Python Libraries and Load ChEMBL Data File
# import libraries import pandas as pd import platform from rdkit.Chem import AllChem as Chem from rdkit.Chem import Descriptors, Descriptors3D, QED, Draw, PandasTools from rdkit.Chem.Draw import IPythonConsole from rdkit import DataStructs print('python version: ' + platform.python_version()) print(f'rdkit version: {rdBase.rdkitVersion}') # ChEMBLからDLしたファイルの読み込み df = pd.read_csv('./2022TYK2.csv',sep=';',)
# output python version: 3.10.6 rdkit version: 2022.03.5
このままdfでデータを開くとわかりますが、数値データの中にstringのNoneがあったり、Smiles stringの中にNanがあったりでpandasの処理を実行できませんでした。
と言うわけで次項のクレンジングが必要になります。
Data Cleansing
# None(string)を0に変換 df = df.replace('None', '0') # datatypeを変更 df['MW'] = df['Molecular Weight'].astype(float) df['pChEMBL Value'] = df['pChEMBL Value'].astype(float) df['AlogP'] = df['AlogP'].astype(float) df['Ligand Efficiendy SEI'] = df['Ligand Efficiency SEI'].astype(float) df['Ligand Efficiendy BEI'] = df['Ligand Efficiency BEI'].astype(float) df['Ligand Efficiendy LE'] = df['Ligand Efficiency LE'].astype(float) df['Ligand Efficiency LLE'] = df['Ligand Efficiency LLE'].astype(float) df['Document Year'] = df['Document Year'].fillna('2025').astype(int) df['#RO5 Violations'] = df['#RO5 Violations'].astype(int) df['Molecule Max Phase'] = df['Molecule Max Phase'].astype(str) # SmilesカラムのNanを空に変換 df['Smiles'] = df['Smiles'].fillna('')
AlogPとか計算できなかった場合Noneが入ってしまってるらしく、何かしらの値を入れないとvisualizationとかできません。
今回は0でreplaceしました。なので、可視化した時など値が0のものは注意が必要です。
Document Yearが入ってないものも時々あったりするんですが、わからないことがわかればいいので2025でreplaceしました。こちらも可視化した時にそういう理解が必要です。
(他にいい方法があったら教えてください)
構造式と骨格情報の追加
# SMILESからMol fileの追加 PandasTools.AddMoleculeColumnToFrame(df,'Smiles','Molecule_Image',includeFingerprints=True) # Murcko Scaffoldの追加 PandasTools.AddMurckoToFrame(df, molCol='Molecule_Image', MurckoCol='Murcko_SMILES') PandasTools.AddMurckoToFrame(df, molCol='Molecule_Image', MurckoCol='Murcko_generic_SMILES', Generic=True) PandasTools.AddMoleculeColumnToFrame(df, molCol='Murcko_Mol', smilesCol='Murcko_SMILES') PandasTools.AddMoleculeColumnToFrame(df, molCol='Murcko_generic_Mol', smilesCol='Murcko_generic_SMILES') # 立体構造の追加 # df['AddHs'] = ddf['Molecule_Image'].apply(lambda x : AllChem.AddHs(x)) # df['3D'] = ddf['AddHs'].apply(lambda x : AllChem.EmbedMolecule(x, AllChem.ETKDGv3())) # df['3D'] = ddf['AddHs'].apply(lambda x : AllChem.RemoveHs(x)) # df['NPR1'] = ddf['AddHs'].apply(lambda x : Descriptors3D.NPR1(x)) # df['NPR2'] = ddf['AddHs'].apply(lambda x : Descriptors3D.NPR2(x))
SmilesをMol filesにしておくとDataFrameのoutputだったりDraw.MolToImageで構造式を描画できます。
PandasToolsは.apply()いらないから楽でいいですね。
文献数と骨格情報のメモ
# 文献数 print("the number of Document ChEMBL ID is " + str(len(df['Document ChEMBL ID'].unique()))) # the number of Document ChEMBL ID is 134 # 骨格数情報 SMILESじゃないとなぜか反映されない print("the number of Murcko generic scaffold is " + str(len(df['Murcko_generic_SMILES'].unique()))) # the number of Murcko generic scaffold is 419 print("the number of Murcko scaffold is " + str(len(df['Murcko_SMILES'].unique()))) # the number of Murcko scaffold is 651
どれくらい文献があって、SARが展開されてきたか、、、これだけじゃわからないので可視化したいですね。。。
Descriptorを追加
# descriptorの追加 df['TPSA'] = df['Smiles'].apply(lambda x : Descriptors.TPSA(Chem.MolFromSmiles(x))) df['Ct HA'] = df['Smiles'].apply(lambda x : Descriptors.HeavyAtomCount(Chem.MolFromSmiles(x))) df['Ct F'] = df['Smiles'].apply(lambda x : x.count('F')) df['HBD'] = df['Smiles'].apply(lambda x : Descriptors.NumHDonors(Chem.MolFromSmiles(x))) df['HBD_Ro5'] = df['Molecule_Image'].apply(lambda x : rdMolDescriptors.CalcNumLipinskiHBD(x)) df['HBA'] = df['Smiles'].apply(lambda x : Descriptors.NumHAcceptors(Chem.MolFromSmiles(x))) df['HBA_Ro5'] = df['Molecule_Image'].apply(lambda x : rdMolDescriptors.CalcNumLipinskiHBA(x)) df['Ct RB'] = df['Smiles'].apply(lambda x : Descriptors.NumRotatableBonds(Chem.MolFromSmiles(x))) df['Ct Ar'] = df['Smiles'].apply(lambda x : Descriptors.NumAromaticRings(Chem.MolFromSmiles(x))) df['Fsp3'] = df['Smiles'].apply(lambda x : Descriptors.FractionCSP3(Chem.MolFromSmiles(x))) df['QED'] = df['Molecule_Image'].apply(lambda x : QED.qed(x))
RDkitを使ってSmilesからDescriptor追加してみました。気が向いたら使ってみます。
df['new column'] = df['target column'].apply(lambda x : f(x))で計算カラムを追加するやり方はだいたいいつも忘れちゃいます。。。
こちらを参考にしてもらうといいかもです。
Summary of Data Table
df[df['Molecule Name'].notna()].filter(items=[ 'Molecule Name', 'Molecule ChEMBL ID', 'Molecule_Image', 'Molecular Weight', 'pChEMBL Value', 'AlogP', 'Ligand Efficiency LLE', '#RO5 Violations', ]).head(3)
名前がついてるような化合物は上市薬だったりツールだったりで色々情報が多いと期待されるので、どんな感じか要約テーブルで見てみます。
df.filter(items=[column name])でカラムの順番を入れ替えれるのも地味に便利でした。

以上、とりあえずChEMBLからとってきたデータを解析できる状態に加工するところまででした。 KNIMEでも同じことはできるんですが、VScodeとかJupyterの方が早いし見やすいので気に入ってます。
次回に続きます