DiffDock@Colabで遊んでみよう
最近diffusionなんとかで色々オープンソースソフトウェアが報告されてます。
DiffDockというドッキングツールもその一つで、git hubやgoogle colabを使って試してみることができます。
いやはや凄い時代ですよね。
そんなわけでミーハーとしてはGoogle colabで早速遊んでみました。
(M1 Macでは色々あって私では動かせませんでした。)
試したこと
Google Colabは細かい設定できなかったので、これまで自分が見たことある複合体8個ほどについてRedockしてみました。
今回はこちらを参考にPDB : 7VU6を紹介してみようと思います。
結果まとめ
- 6/8はオリジナルと同じ結合ポケットを予測
(ただし予測結果が40個出力され、その全てが正しいわけではない) - 4/8はDocking Poseも近い。置換基の位置関係が揃っているくらいの感じ。
- 2/8は相互作用も再現した。これは見事。
- 結晶構造の2量体間隙にDockingされたものもあった(artifact?)
- 水を考慮してDockingしていた。深層学習でこれを考慮するのは非常に興味深い。
SARS-CoV2 3CL protease
実は以前このブログでも取り上げていました。最近は治療薬のニュースで非常にホットな標的と言っていいでしょう。
この3CL proteaseのタンパクは確か2量体で機能するんですが、結晶構造の2量体もおそらく同様の構造をとっているのではないでしょうか。
chian Aを赤、chain Bをピンクで示しました。なんかハート型に見えるところがなんとも悍ましいというか忌々しいというか不思議な見た目です。
 緑で示したEnsitrelvirをはじめとする治療薬がコロナウイルスの楔になるの乞うご期待ですね。
緑で示したEnsitrelvirをはじめとする治療薬がコロナウイルスの楔になるの乞うご期待ですね。

あ、HUNTER x HUNTER再開楽しみです。
Docking site
Defaultのsettingだと40poseが表示されます。
最初のESM(何なのかはわかってませんすみません)作成に少し時間がかかるくらいで、2回目以降は読み込むPDBを変えても非常に計算が早かったです。さすがはGPU。
よくあるDocking ProgramのGlideやVinaはDockingする範囲をGridで指定することが多いと思いますが、DiffDockはGlobal searchするみたいです。
2 min/40 pose/ligandなので、体感的にはGlideやVinaより早い印象です(マシンスペックが違うので実際比べると違うかもしれませんが)。
結果は下記の通りで、オリジナルの位置とほぼ相違なく収束しました。

 他の結晶構造では、
他の結晶構造では、
- オリジナルを再現しないものも2例ありました。
- 7VU6では見られませんでしたが、2量体間隙にdockingされるケースもありました。
- rank 1が必ずしも正解するわけではありませんでした。
Docking pose
緑がオリジナル、水色がrank1のposeです。
母核、置換機の位置は概ねあっていると言えそうです。
rank10くらいまではいい感じの結果でしたが、以降はposeが外れてるものも増えてくる感じでした。
他の結晶構造では正解poseが一つも含まれないものもありましたが、そういう場合のRMSDがどんな値になるか気になりました。

Ligand-Receptor Interaction
7VU6では完璧な再現はできませんでしたが、相互作用まで再現した結晶構造も2つありました。
あんな短時間のGlobal search (じゃないのかもしれませんが)で辿り着けるのは脱帽でした。
興味深い・気になった点として、
- ここまでposeを再現したにも関わらず相互作用距離に届かないものが結構多い
(つまり相互作用を計算しているわけではない。そもそもそういうコンセプトか) - 水もDockingの対象になっている
(今回は未設定なので、考慮すれば精度は上がるかも)

他にはKdの強弱が精度とどれくらい相関しそうかとか、気になることは尽きないなーと思いました。
DiffDockといいColabといいAlphaFold2といい、いやーほんと凄い時代ですね。
むしろwetの需要が上がるよね?って違うのかな?
おしまい。
TeachOpenCADDを改造するの巻
Grüezi,
先日ナイメストさんの記事でPDBのNodeが復活していることを知りました。
【W8】タンパク質データの取得_05_Step2_02_Query2RCSB|ナイメスト @KNIME9 #note #一歩踏みだした先に https://t.co/gwJPdg9q0e
— 巨人の肩の上でKNIMEを語る (@Note_of_KNIME) August 12, 2022
と言っても完全復活ではなく、RSCB PDBの現行APIに合わせて動くようにしましたって感じでしょうか。開発のVernalisさんも困惑を隠せない雰囲気ですし。
そんなわけで、TeachOpenCADDのW8の流れでPDBを取得~って言うのはわかるんですが、実際に使う身としてはresolution top 4しか見ないってことはあまりない気がしました。
 最近の私の好みで言うとポケットごとにどんなリガンドが報告されているか知りたいんですが、KNIMEでどう処理するかは気が向いたら勉強します。とりあえずは過去にやっていたように、どんなリガンドが報告されているか、それを分類したらどうなるか、辺りが気になりますが、このWorkFlowだとKNIME上ではリガンドの構造が出てきません。
最近の私の好みで言うとポケットごとにどんなリガンドが報告されているか知りたいんですが、KNIMEでどう処理するかは気が向いたら勉強します。とりあえずは過去にやっていたように、どんなリガンドが報告されているか、それを分類したらどうなるか、辺りが気になりますが、このWorkFlowだとKNIME上ではリガンドの構造が出てきません。
なくても全然支障はないけど
めんどうだからってつけない人もいるよ
どうする?
構造あったら見るよね?
でもcustom report作ってくれるNodeがAPI使用変更でなくなったんだよね...
それ、APIで取れます。
っていうのが今日のアルトコロニーの定理です (出典はググってね
というわけで本日は勝手に「STEP 5:リガンドとAbstractを取ってくる」です。
 STEP 3に入るとtop 4に絞られちゃうので、STEP 2のSorterから引っこ抜いてきます。
STEP 3に入るとtop 4に絞られちゃうので、STEP 2のSorterから引っこ抜いてきます。
構造を足した後に全部ダウンロードするか、別の要件でfilterするかは好みで判断してください。
2022.8の時点で178構造がHitしているので、構造ごとにClusteringしたりしないと見る気が失せる気がします。
- Overview
- Column Expression ~Ligand information~
- GET Request > JSON to Table
- Column Filter
- Column Splitter
- Column List Loop Start
- Molecular Type Cast
- Loop End (Column Append)
- Joiner
Overview
こんな感じです。
 すでにPDB IDとLigand IDが取得してあるので、そのまま流用する形でWorkFlowを作ります。作成者さんありがとう。
すでにPDB IDとLigand IDが取得してあるので、そのまま流用する形でWorkFlowを作ります。作成者さんありがとう。
Column Expression (String Manipulationsでもいいけど)でURLを作成して、GET RequestでJSON取得、Tableに分解してSMILESのStringsをMolecule Type Castでchemical structureに変換します。
今回初めて作ったのですが、複数(4カラム)含まれているSMILESのどれを採用すればいいのかわからなかったので、Column List Loopで手当たり次第に化学構造に変換して目視確認しました。一旦通れば必要なカラムだけ変換でいいと思います。
Column Expression ~Ligand information~
まず始めにですが、TeachOpenCADDではGraphiQLを使ってAPI Queryを作成してますが、私はさっぱり記述法が理解できませんでした(爆
Web Services Overview
https://data.rcsb.org/graphql/index.html
というわけで、REST APIで許してください。こちらを参考にLigandやPDBのIDを使ってURLを作成しています。

 結果はこんな感じですね。
結果はこんな感じですね。

GET Request > JSON to Table
生成したURLにGET RequestするとJSONが返ってきます。

 JSONだけ見てもここでは意味不明なので、JSON to Tableを使って各要素をtableに分解してやります。設定はデフォルトです。
JSONだけ見てもここでは意味不明なので、JSON to Tableを使って各要素をtableに分解してやります。設定はデフォルトです。
 下のようなtableになって返ってきますが、
下のようなtableになって返ってきますが、descriptorの部分が構造式情報のようです。ただし、SMILESとInChI Keyが混ぜこぜです。よくわからないのでSMILESが入ってるdescriptor、descriptor (#1)、descriptor (#3)、descriptor (#5)だけをあとでfilterして残します。

構造情報は取れたので、ついでにPubmedのabstractとキーワード、doiあたりもAPIで取ります。URLは前項で生成済みなので、同じくGET RequestとJSON to Tableで情報を取得します。

JSON to Table するとこんな感じ。
 感の良い方はGET Request x2してからまとめてJSON出来ないの?って思うかもしれません。結論から言うと上手く出来ませんでした。設定の問題なのかもなので、分かる方は教えてください。
感の良い方はGET Request x2してからまとめてJSON出来ないの?って思うかもしれません。結論から言うと上手く出来ませんでした。設定の問題なのかもなので、分かる方は教えてください。
Column Filter
情報量が多いので使うカラムだけ残します。

Column Splitter
descriptorのカラム4つ分はMolecular Type CastでStringsからSMILESに変換したいです。何個もNodeを置いてもいいですが、同じ処理を複数カラムにかける場合はLoopを使えます。
Column SplitterでLoopの対象カラムをSplitしておきましょう。

Column List Loop Start
split済みなので設定不要な気がしますが、こいつらにNodeで同じ処理します。

Molecular Type Cast
StringsをSMILESにするNodeなわけなんですが、Variablesのcolumn nameをcurrentColumnNameにしておきます。これによってカラムが変数になります(多分

Loop End (Column Append)
設定不要です。Loopが終了します。
結果はこんな感じです。
カラム名を右クリックしてrendererを2D depictionに変更します。
rowが小さくてめっちゃ見づらい場合はviewのRow Heightを300くらいにしてやります。

Joiner
諸々全部くっつけておしまいです。
ざっと見た感じでほとんどがkinase pocketのbinderですね。clustringして実際の結晶構造の結合様式とかと比べてみたら面白いかもしれません。気が向いたらやってみようかな。
おしまい。
探してみた ~PyMOLで相同性の高いmotifの探し方~
先日JAK familyの相同性、というか配列の相違点?、を興味本位でPyMOLを使って紹介したところ、
PyMOLで相同性を見てみました。
— おじさん (@snowborderjack) August 1, 2022
python使ってもっとスマートに仕事したいなー。
はてなブログに投稿しました #はてなブログ
PyMOLで相同性を確認してみた - 或る化みす途のブログhttps://t.co/1iwquWR9pk
KNIMEでは勝手に師事しているすさんにPyMOLでもご指導頂けました。
今日は有給。ひさーしぶりにブログを書いたよ。 #はてなブログ #PyMOL
— す (@sumtat_) August 5, 2022
【PyMOL】どこが違うのかな? - 非プログラマーのためのインフォマティクス入門。(仮)https://t.co/SvBVGpePYk
地味に嬉しかったので、私が使ってるPyMOLのfindseqというscriptも紹介します。
すさんの紹介してくれたcolor_by_mutationと違ってダウンロードできる形のスクリプトが画面右側に用意されているので、findseq.pyのかたちでダウンロードすればOKです。
PyMOL上でのactivateの仕方はすさんの方法に準じるので割愛します。
今回の題材は同じくJAKiのdelgocitinib@JTというアトピーの塗り薬です。
https://pubs.acs.org/doi/full/10.1021/acs.jmedchem.0c00450
PDB idは7C3Nです。前回のpfizerのJAKiの6bbuと併せてfetchで読み込んでおきましょう。全然重なってないのでalignもかけます。
fetch 6bbu 7c3n align 6bbu, 7c3n, object=aln color_by_mutation 6bbu, 7c3n hide lines hide sticks select ar7, byres organic around 7
水分子は鬱陶しいので隠してあります。 選択性に関係してそうなのはp-loopのあたりでしょうか。
 余計なリボンを消してよーく見てみると、delgocitinibのシアノ基はかろうじて変異近傍の主鎖を捉えてます。反対にpfizerのcompound 25は少し浮いているp-loopに対してEt基で空間を埋める程度のお仕事でしょうか。両社ともお見事。
余計なリボンを消してよーく見てみると、delgocitinibのシアノ基はかろうじて変異近傍の主鎖を捉えてます。反対にpfizerのcompound 25は少し浮いているp-loopに対してEt基で空間を埋める程度のお仕事でしょうか。両社ともお見事。

ところで私のようなkinaseのど素人は、俗に言うDFG motifとかがどの辺でどんな仕事してるか
全くわかりません(勉強不足ですみません)。
そんな時に使えるのが今日紹介するfindseqです。
findseq DFG, enabled, DFG enable aln
 対象objectが一つであれば
対象objectが一つであればDFGのmotifだけがselectされるはずなんですが、複数のobjectで一度に処理すると違うmotifも認識されちゃってますね。。。
alnのobjectでsequence alignmentがかかっているので、赤枠のように同じ位置のDFGがいわゆるDFG motifだと思われます。
show sticks, DFG
 赤枠の
赤枠のsticksのあたりがDFG motifですが、先ほどの選択性に関わっていたp-loopと比べて位置情報レベルでも相同性が恐ろしく高いですね。
こんな感じで、resnでは出来ない配列検索ができます。良い子のみんなはこれがなんで便利なのかわかるよね?
以上、知らないおじさん同士の知的なSNSでのやり取りの仕方を探してみました。おしまい。
PyMOLで相同性を確認してみた
JAKiってキナーゼのわりには構造がシンプルでも活性と選択性が出てそうで、ケミストはとっつきやすいのかもですね!
— へい🍅 (@HiGoing) July 3, 2022
ほんまかいな、ということで今回はPyMOLがインストールしてあれば誰でも出来る簡単な相同性の確認方法です。
お題の報告はこちらです。
https://pubs.acs.org/doi/10.1021/acs.jmedchem.7b01598
PDBという単語で検索をかけると最後の方に論文で登録されたPDB IDの6BBUと6BBVが見つかると思います。
PyMOLを開いたらfetchでloadしましょう。
fetch 6bbu 6bbv

全然重なってないのでalignします。
align 6bbu, 6bbv
6bbuを6bbvの場所に持っていってalignするよってコマンドです。
 いい感じに重ね合わさってればOKです。
いい感じに重ね合わさってればOKです。
次にリガンド周辺だけ抜き取ります。
create u, byres organic expand 10 and 6bbu
create v, byres organic expand 10 and 6bbv
disable 6bbu or 6bbv
 そして改めてalignmentのobjectを作ります。
そして改めてalignmentのobjectを作ります。
align u, v, object=aln
set seq_view, 1
もしくは画面右下のSをクリック
 このsequenceは構造をもとにalignmentされています。つまり重なっている残基同士が並んでいる状態です。
このsequenceは構造をもとにalignmentされています。つまり重なっている残基同士が並んでいる状態です。
alnのobjectをinactiveにしてやることで解除することもできます。
このsequenceを見ながら一致してない残基を見つけてやればいいわけです。
(私のcoding能力では自動抽出がまだできませんごめんなさい)

show sticks, sele
zoom byres orgnic around 3

リガンド周辺に選択性が出せそうなアミノ酸残基との相互作用が見当たりません...。変態ですか。誰ですかJAKはまだ頑張れるなんて呟いた人は。
久々に頑張ったので夏休みに入りたい気持ちでいっぱいです。皆さんも体調にお気をつけて。
~終わり~
-追記-
ちなみにこのalnのobjectはFASTAやCLUSTALで保存もできるので普通の配列解析もできるはずです(私ができないだけで)
#CLUSTAL形式 save aln.aln, aln #FASTAA形式 save aln.fasta, aln
CLUSTAL形式はterminalからcatコマンドで確認してやってもいいと思います。
cat aln.aln

~さらに追記~ すさんからご助言いただきまして、windowsは下記のscriptを実装できました。
pymolwikiの"Color By Mutations"を使うと、ちょっと楽できます。https://t.co/UytFMKB8CQ
— す (@sumtat_) August 2, 2022
Macで動かないのはなんでなん?dependency errorとか?
この辺の便利ツールは.pymolrcに書くとカオスなので、xxx.pyでscriptにしてimport xxxくらいが散らからないかなと思いました。もっとシンプルに自動化する方法もあるけど、使用頻度が低すぎて忘却必須ですね。
-報告- 拙い記事をすさんにフォローアップいただきました。
今日は有給。ひさーしぶりにブログを書いたよ。 #はてなブログ #PyMOL
— す (@sumtat_) August 5, 2022
【PyMOL】どこが違うのかな? - 非プログラマーのためのインフォマティクス入門。(仮)https://t.co/SvBVGpePYk
.pymolrc
PyMOLの自作.pymolrcファイル
#2022.6.29
リガンド周りのラベリングをスッキリさせました。
#2022.8.10
AlphaFold2のplDDTスコアのカラーリングをラボォさんのtweetを参考にalias化しました。
tagをシンプルな設計に変えました。
FASTAをprintするalias作りました。
maestroっぽいセレクタのalias作りました。
そのほか細々変えました。
#set cartoon_transparency, 0.1
#set transparency, 0.4
set surface_color, white
set sphere_scale, 0.4
set label_position, (3,1,1)
set label_size, 25
# set ray_trace_mode, 1
set ray_shadow, off
bg_color silver
one_letter ={'VAL':'V', 'ILE':'I', 'LEU':'L', 'GLU':'E', 'GLN':'Q', \
'ASP':'D', 'ASN':'N', 'HIS':'H', 'TRP':'W', 'PHE':'F', 'TYR':'Y', \
'ARG':'R', 'LYS':'K', 'SER':'S', 'THR':'T', 'MET':'M', 'ALA':'A', \
'GLY':'G', 'PRO':'P', 'CYS':'C'}
alias al, alignto; zoom
alias sp, split_chains enabled; alignto; zoom
alias cc, util.color_chains("(enabled and name CA)",_self=cmd)
alias pl, preset.ligand_cartoon("enabled",_self=cmd);show spheres, solvent
alias see, zoom byres organic around 8 and chain A
alias del, as nb_spheres, solvent
alias sol, as nb_spheres, solvent; hide nb_spheres
alias line, show lines, byres organic around 4 and enabled
alias hb, select ligand, organic and enabled; cmd.dist("contact","(ligand)","(byobj (ligand)) and (not (ligand))",quiet=1,mode=2,label=0,reset=1); cmd.enable("contact")
alias well, show surface, byres organic expand 5 and enabled
alias 9, create 9, byres organic expand 9 and enabled; disable !9; see
alias 5, create a5, byres organic expand 5 and enabled; disable !5; see
alias x, select sele, byres sele expand 4 and enabled
alias tag, label n. CA and sele, "%s%s" % (one_letter[resn], resi)
alias untag, hide label, sele
alias k, hide sticks, polymer.protein; show sticks, resn lys
alias c, hide sticks, polymer.protein; show sticks, resn cys
alias s, hide sticks, polymer.protein; show sticks, resn ser
alias h, hide sticks, polymer.protein; show sticks, resn his
alias t, hide sticks, polymer.protein; show sticks, resn thr
alias r, hide sticks, polymer.protein; show sticks, resn arg
alias d, hide sticks, polymer.protein; show sticks, resn asp
alias e, hide sticks, polymer.protein; show sticks, resn glu
alias n, hide sticks, polymer.protein; show sticks, resn asn
alias q, hide sticks, polymer.protein; show sticks, resn gln
alias y, hide sticks, polymer.protein; show sticks, resn tyr
alias f, hide sticks, polymer.protein; show sticks, resn phe
alias w, hide sticks, polymer.protein; show sticks, resn trp
alias g, hide sticks, polymer.protein; show sticks, resn gly
alias a, hide sticks, polymer.protein; show sticks, resn ala
alias m, hide sticks, polymer.protein; show sticks, resn met
alias p, hide sticks, polymer.protein; show sticks, resn pro
alias fasta, print(cmd.get_fastastr('enabled'))
alias plddt, spectrum b, rainbow_rev, maximum=100, minimum=25
外部デバイスから取り込んだファイルのアクセス権を一括で変更するコマンド
フィルムカメラで撮った写真を現像した時にデータファイルのCD-Rをもらう、みたいなやや時代遅れなことをいまだにやってます。
そんな時、データをMacにただコピペしただけではアクセス権がRead Onlyになっている時があります。
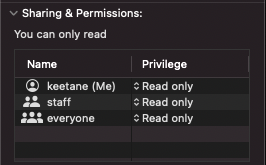
このままだとiPadに転送したり他のデバイスにコピーしたりができないんで、ファイルごとにRead&Writeに変更する必要があります。手作業でやったら大変です...
そんな時はTerminalからcdでWorkin directoryに移動してこのコマンドを使います。
cd ~/Picture chmod -R 755 <directory name>
file nameをまとめて処理したい場合は、directoryをもう一つ下げてwild cardを使います。オプションの-Rも外します。
wild cardだと特定の拡張子だけを指定することも出来たりしますね。
cd ~/Picture/directory chmod 755 *.JPG
755の意味は覚えれないのでこちらを参照ということで。
Gitで変更点を見るdiffとかstatusのいろいろ
diffとかstatusとかいつも忘れるのでメモ
git status
変更点やstaging area, commitとの変更点を要約表示
git diff --stat
git diffだと変更点全てが表示されて把握しづらい。 まずはどれくらい変更したかをgit diff --statで確認。
git diff --name-only
どのファイルが変更されたかだけなら--name-onlyでいい。
git diff --staged (--cached)
staging areaとHEADの差分を確認
git diff branchA..branchB
ブランチ同士の変更点を見る。
--statや--name-onlyも一緒に使える。
git diff HEAD^
前回のコミットと今回のコミットの差分を見る。
git diff remote name/branch name..HEAD
新しくdummyというfileをローカルに追加したとすると
git diff origin..HEAD --stat # dummy | 1 +
のように結果が返ってくる。 originからHEADでは変更が加えられましたよ、反対に
git diff HEAD...origin --stat # dummy | 1 -
HEADから見てoriginはファイルが1つ少ないよということ。
コード同士を比較しないと何も思い出せませんな。